protein-visualizer
A free and open-source program to visualize proteins and the function of its individual amino acids through deep learning.
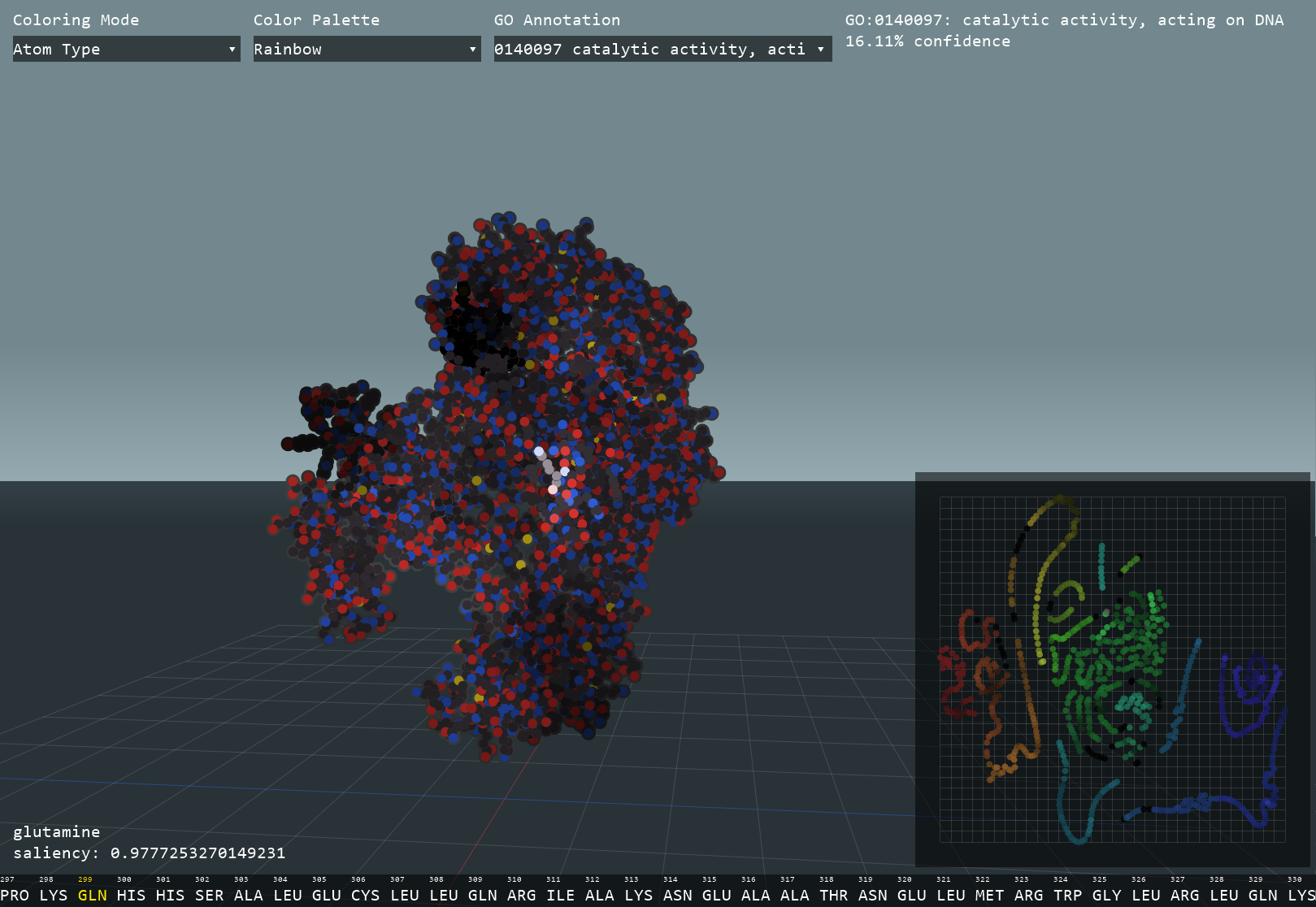
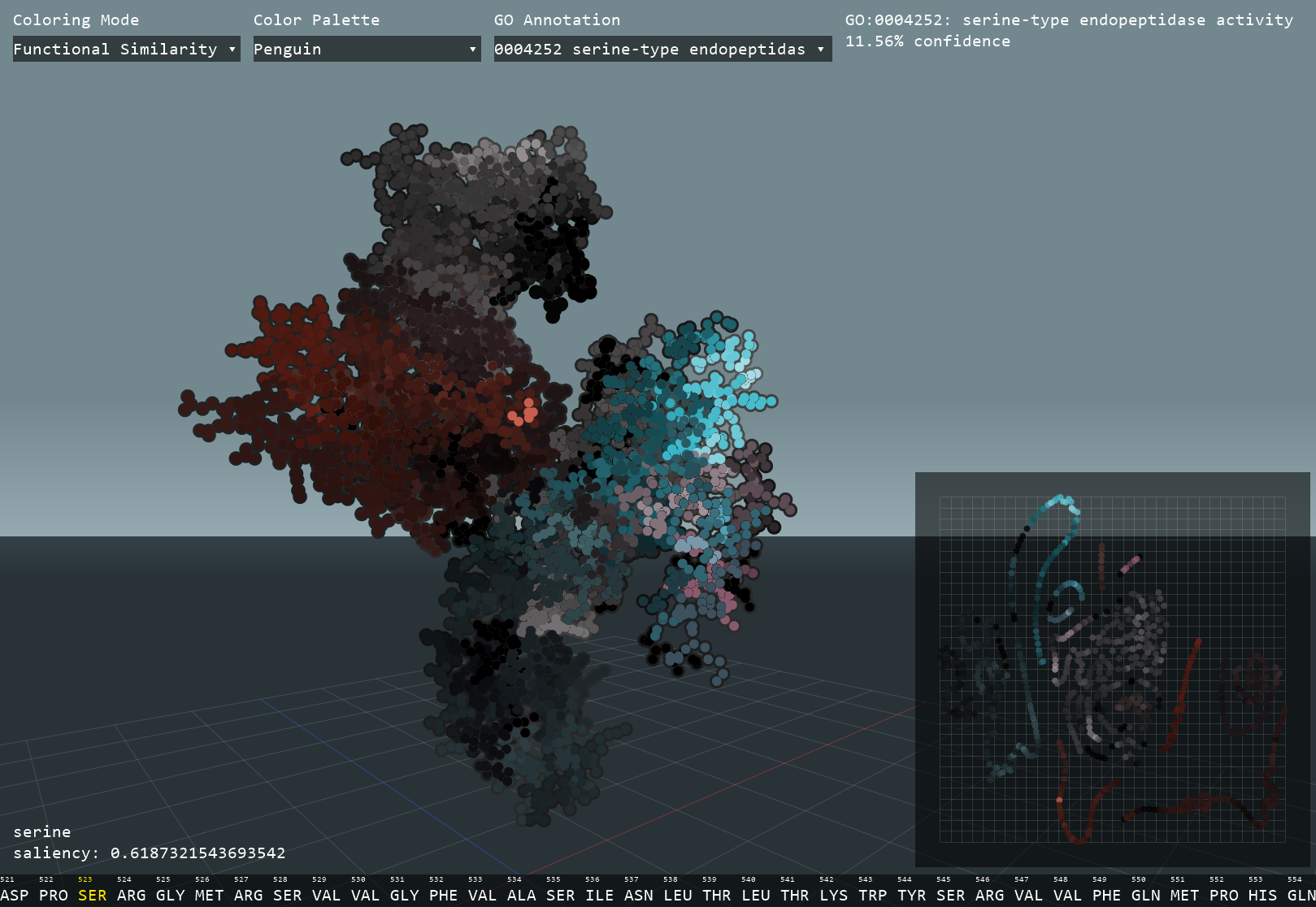
How does it work?
This interactive visualizer uses a neural network to identify functional and structural clusters within a protein by calculating embeddings, numerical expressions of each amino acid. The high-dimensional data is then transformed into 2D in order to easily interpret the possible functional components of a protein. Next, a graph convolutional network uses both the 3D structure and amino acid sequence to predict Gene Ontology (GO) annotations that describe the molecular function of the protein. Through Gradient-weighted Class Activation Mapping (GradCAM), the program can calculate how much each amino acid contributed to each GO prediction and subsequently allow for the identification of functional residues.
This program was written entirely with Python, using Pyglet as an OpenGL interface.
Disclaimer: All of the data (except for 3D structure) shown in this program is generated through machine learning and thus cannot be verified to be completely accurate. It is recommended to double-check the predicted functions through experimental sources
Usage
Installation/Running
Download and extract the latest release from the
protein-visualizer release page.
Run protein-visualizer-x.x/main.exe and wait for the help terminal to appear.
Input
When the program is loaded, it will prompt you for a protein file in either the Protein Data Bank (.pdb) format or ModelCIF (.cif) format. The RCSB Protein Data Bank is a great source to find proteins to test out with this program. If your file has more than one amino acid chain, the help terminal will prompt you to type the name of the chain you want to render. After some time computing predictions about your protein, the interactive interface will open with your protein ready to view.
Controls
Left Mouse: Translate the camera and navigate the embedding space.Right/Middle Mouse: Rotate the camera.Up/Down Arrow: Increase/decrease atom point size.O: Toggles atom outline.
Using Generated Data
Before viewing a protein, the program caches amino acid embeddings and Gene Ontology (GO) annotations in
protein-visualizer-x.x/data/[protein_name]_data.json to reduce load times the next time the
program is run with the selected protein.
If you wish to use the prediction data yourself, you can parse through the data file. The following information is stored as a dictionary for each protein:
GO_ids: A list of predicted GO IDs.GO_names: A list of descriptions for each predicted GO ID.confidence: A number between 0 and 1 denoting the confidence of each GO prediction.saliency_maps: A matrix where each row contains numbers between 0 and 1 denoting how strongly an amino acid contributed to the prediction of that row's GO annotation.sequence: The protein's amino acid sequence in the FASTA format (without a header).embedding_points: A flattened list of (x, y) points denoting the coordinates of each amino acid in the latent space after being transformed into 2D.cluster_indices: A list of integers denoting which cluster each amino acid belongs to, with -1 meaning no cluster.
You can also generate the data file without opening the GUI by executing the program from the command line:
protein-visualizer.x.x/main.exe [path_to_pdb_file] [optional_chain_id]. If no chain is selected,
the program will automatically select one rather than prompt the user like when the GUI is opened.
Code
All of the code is available under the MIT license on the protein-visualizer GitHub repository.
Note that in order to run the code, you must download the most recent release of the program and copy the
protein-visualizer-x.x/saved_models/ folder into the main directory.
Feel free to take any part of the code to expand or transform, just make sure you keep the attributions to ProSE and DeepFRI!
File Structure
deep_learning/: Contains ProSE and DeepFRI code that generates amino acid embeddings and GO annotations respectively using the pre-trained models.docs/: Contains this website.gui/: Contains code to draw user interface widgets, such as buttons and the amino acid label.img/: Contains icons used when running the program.renderers/: Contains code used to draw the 3D protein and the 2D embedding space.saved_models/Contains the pre-trained models created by the ProSE and DeepFRI teams. Must be copy and pasted from the latest release due to large size.batch_predict.py: Example script of using the program from the command line.main.py: Entrypoint to the program.protein.py: Contains classes that parse and organize data about a protein.
Attribution
- Eric Alfaro: Primary program developer as part of an MIT UROP project under the guidance of Dr. Manolis Kellis and Dr. Tianlong Chen.
-
Protein Sequence Embeddings (ProSE): Amino acid embedding code and pre-trained models.
- Bepler, T., Berger, B. Learning the protein language: evolution, structure, and function. Cell Systems 12, 6 (2021). https://doi.org/10.1016/j.cels.2021.05.017
- Bepler, T., Berger, B. Learning protein sequence embeddings using information from structure. International Conference on Learning Representations (2019). https://openreview.net/pdf?id=SygLehCqtm
-
Deep Functional Residue Identification (DeepFRI): GO annotation prediction code and pre-trained models.
- Gligorijevic, Vladimir and Renfrew, P. Douglas and Kosciolek, Tomasz and Leman, Julia Koehler and Cho, Kyunghyun and Vatanen, Tommi and Berenberg, Daniel and Taylor, Bryn and Fisk, Ian M. and Xavier, Ramnik J. and Knight, Rob and Bonneau, Richard. Structure-based function prediction using graph convolutional networks. Cold Spring Harbor Laboratory (2019). https://doi.org/10.1101/786236